The MODY-associated KCNK16 L114P mutation increases islet glucagon secretion and limits insulin secretion resulting in transient neonatal diabetes and glucose dyshomeostasis in adults
- Department of Molecular Physiology and Biophysics, Vanderbilt University, United States
- Department of Chemistry, Vanderbilt University, United States
- Center for Stem Cell Biology, Vanderbilt University, United States
- Department of Cell and Developmental Biology, Vanderbilt University, United States
eLife assessment
This study characterizes how a point mutation in the TALK-1 potassium channel, encoded by the KCNK16 gene, causes MODY diabetes. The mutation, L114P, causes a gain-of-function to increase K+ currents and inhibit glucose-stimulated insulin secretion. Increased glucagon likely results from paracrine effects in the islets. The data are convincing and the work will be valuable for understanding islet function.
https://doi.org/10.7554/eLife.89967.3.sa0Significance of the findings:
Valuable: Findings that have theoretical or practical implications for a subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Convincing: Appropriate and validated methodology in line with current state-of-the-art
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
The gain-of-function mutation in the TALK-1 K+ channel (p.L114P) is associated with maturity-onset diabetes of the young (MODY). TALK-1 is a key regulator of β-cell electrical activity and glucose-stimulated insulin secretion. The KCNK16 gene encoding TALK-1 is the most abundant and β-cell-restricted K+ channel transcript. To investigate the impact of KCNK16 L114P on glucose homeostasis and confirm its association with MODY, a mouse model containing the Kcnk16 L114P mutation was generated. Heterozygous and homozygous Kcnk16 L114P mice exhibit increased neonatal lethality in the C57BL/6J and the CD-1 (ICR) genetic background, respectively. Lethality is likely a result of severe hyperglycemia observed in the homozygous Kcnk16 L114P neonates due to lack of glucose-stimulated insulin secretion and can be reduced with insulin treatment. Kcnk16 L114P increased whole-cell β-cell K+ currents resulting in blunted glucose-stimulated Ca2+ entry and loss of glucose-induced Ca2+ oscillations. Thus, adult Kcnk16 L114P mice have reduced glucose-stimulated insulin secretion and plasma insulin levels, which significantly impairs glucose homeostasis. Taken together, this study shows that the MODY-associated Kcnk16 L114P mutation disrupts glucose homeostasis in adult mice resembling a MODY phenotype and causes neonatal lethality by inhibiting islet insulin secretion during development. These data suggest that TALK-1 is an islet-restricted target for the treatment for diabetes.
Introduction
Maturity-onset diabetes of the young (MODY) is a collection of monogenic forms of early-onset familial diabetes resulting from β-cell dysfunction. To date, mutations in 15 genes involved in β-cell development and function have been associated with MODY (Broome et al., 2021). Monogenic diabetes cases account for approximately 1–5% of the total diabetic patient population (Kavvoura and Owen, 2012; Zhang et al., 2021). However, as the phenotype of MODY overlaps with other forms of diabetes, many patients are misdiagnosed (Shields et al., 2010; Greeley et al., 2022). Data from monogenic diabetes registries also suggests that in the US, ~70% of the registered cases still do not have a known genetic cause, and similarly many patients in the UK (~50%) with monogenic diabetes have MODY-causing mutations that remain to be determined (Bowden et al., 2021; Shepherd et al., 2016). Additionally, some MODY-associated mutations are only reported in single families, and thus require genetic and mechanistic confirmation (Yamagata et al., 1996; Pearson et al., 2005). We recently identified a mutation in KCNK16, the gene encoding TALK-1 channels (p. Kcnk16 L114P), which co-segregates with MODY in a four-generation family (Graff et al., 2021). As KCNK16 is the most islet-restricted and abundant β-cell K+ channel transcript, the Kcnk16 L114P mutation is likely to perturb β-cell function and cause MODY (Bramswig et al., 2013; Blodgett et al., 2015). This was further strengthened by a recent report of a Japanese family with the identical KCNK16 mutation (p. Kcnk16 L114P) segregating with MODY (Katsuyuki Matsui et al., 2023). Thus, it is important to determine the mechanistic underpinnings of how this specific mutation in TALK-1 impacts islet function and results in glucose dyshomeostasis.
TALK-1 is a key regulator of β-cell membrane potential (Vm), glucose-stimulated Ca2+ influx, and insulin secretion (Vierra et al., 2015). A non-synonymous gain-of-function polymorphism in KCNK16 (rs1535500; p. Kcnk16 A277E) is also associated with an increased risk for type 2 diabetes (T2D) (Cho et al., 2011; Mahajan et al., 2014). Moreover, we recently determined that the MODY-associated Kcnk16 L114P mutation results in a significant gain-of-function in TALK-1 K+ flux (Graff et al., 2021). Thus, there is strong genetic evidence that alterations in TALK-1 function (e.g. Kcnk16 L114P and Kcnk16 A277E) result in diabetic phenotypes. Interestingly, when heterologously expressed in β-cells, TALK-1 L114P inhibited glucose-stimulated membrane potential (Vm) depolarization and Ca2+ influx in most β-cells. Although TALK-1 L114P-mediated changes in inhibition of electrical activity and Ca2+ influx would be predicted to fully inhibit glucose-stimulated insulin secretion, they only resulted in partial blunting of β-cell insulin secretion. However, this is consistent with Kcnk16 L114P MODY patients that required low-dose insulin therapy which also suggested that this mutation does not fully suppress glucose-stimulated insulin secretion. This differs from previous studies showing that inhibition of β-cell Ca2+ influx with either K+ channel pharmacological activation or gain-of-function mutations (e.g. KATP R201H) results in complete inhibition of glucose-stimulated insulin secretion (Gloyn et al., 2004). Therefore, it is critical to determine if endogenous TALK-1 L114P expression also shows complete inhibition of β-cell Ca2+ influx and how this impacts glucose-stimulated insulin secretion. Another potential mechanism for the modest impact of Kcnk16 L114P on insulin secretion could be due to its function in other islet cells. For example, TALK-1 is also expressed in δ-cells where its activity limits somatostatin secretion (Vierra et al., 2018). Because somatostatin exerts inhibitory tone on islet β- and α-cells, TALK-1 L114P-mediated reductions in δ-cell somatostatin secretion would be predicted to increase both glucagon and insulin secretion. Due to the β-cell-intrinsic role of TALK-1 L114P channels, the effect of somatostatin on glucose-stimulated insulin secretion might be limited; however, it remains to be determined if the fasting hyperglycemia observed in KCNK16-MODY patients is due in part to elevated glucagon secretion.
The nature and severity of MODY phenotypes is dictated by how specific gene mutations affect β-cell function. KATP is the only ion channel besides TALK-1 to be linked to MODY; this is due to mutations in genes encoding the KATP channel complex (KCNJ11 and ABCC8) or mutations in genes affecting ATP synthesis (e.g. glucokinase) (Bonnefond et al., 2012; Huopio et al., 2003). Interestingly, MODY-associated mutations in KCNJ11 and ABCC8 have also been found to cause other diabetic phenotypes including permanent or transient neonatal diabetes, and late-onset diabetes (Yorifuji et al., 2005; Devaraja et al., 2020). Although the two families with Kcnk16 L114P exhibit a MODY phenotype, it remains to be determined if KCNK16 mutations are associated with other diabetic phenotypes besides MODY. Indeed, it was originally predicted that mutations that cause a substantial gain-of-function in TALK-1 channel activity (e.g. Kcnk16 L114P) would act similarly to the gain-of-function mutations in KATP subunits that cause neonatal diabetes (Vierra et al., 2015). However, the biophysical activity of TALK-1 channels differ in many ways from KATP channels, and this could contribute to more modest MODY phenotype observed in patients with KCNK16 L114P. For example, depolarization-dependent activation (outward rectification) of TALK-1 currents may limit the activity of TALK-1 L114P channels at resting Vm (Girard et al., 2001). KATP channels also show larger unitary conductance than TALK-1, not much voltage dependence, and are nucleotide gated (Rorsman and Ashcroft, 2018; Kang and Kim, 2004). The unique biophysical properties of KATP and TALK-1 channels predict mechanistic differences in their corresponding gain-of-function phenotypes. However, it remains to be elucidated how endogenous Kcnk16 L114P disrupts islet electrical activity and glucose homeostasis.
Here, we developed a mouse model harboring the MODY-associated Kcnk16 variant p. Kcnk16 L114P to investigate the impact of this mutation on glucose homeostasis and confirm its association with MODY. Interestingly, we observe increased neonatal lethality in heterozygous and homozygous Kcnk16 L114P mice in the C57BL/6J (B6) and the B6;CD-1 (ICR) mixed genetic backgrounds, respectively, likely due to severe hyperglycemia and lack of glucose-stimulated insulin secretion. Whereas in adult mice, Kcnk16 L114P blunts glucose-stimulated β-cell electrical activity, Ca2+ handling, and glucose-stimulated insulin secretion, thus significantly impairing glucose tolerance. These data strongly suggest that alterations in TALK-1 activity can disrupt islet hormone secretion and glucose homeostasis. Importantly, this study further confirms that the Kcnk16 L114P mutation results in a MODY phenotype, but additionally predicts that gain-of-function TALK-1 mutations cause transient neonatal diabetes.
Results
Kcnk16 L114P neonates exhibit loss of glucose-stimulated Ca2+ entry and insulin secretion leading to transient neonatal hyperglycemia and death
To confirm the association of Kcnk16 L114P in causing MODY, a mouse model harboring the Kcnk16 mutation was developed in the C57BL/6J background using CRISPR/spCas9 (B6 Kcnk16 L114P; Figure 1A and Figure 1—figure supplement 1A and B). Surprisingly, heterozygous Kcnk16 L114P (L/P) mice exhibited neonatal lethality as indicated by extremely low number of Kcnk16 L114P (L/P) mice at weaning (Figure 1B). To increase the likelihood of survival of neonates, B6 Kcnk16 L114P (L/P) mice were crossed with outbred CD-1 (ICR) mice resulting in a progeny in the mixed (50:50) background (B6;CD-1 Kcnk16 L114P; Figure 1A). Intriguingly, in the mixed background, neonatal lethality was observed in the homozygous B6;CD-1 Kcnk16 L114P (P/P) mice on ~postnatal day 4 (P4), but not in the heterozygous B6;CD-1 Kcnk16 L114P (L/P) mice (Figure 1—figure supplement 1C). Lethality was likely independent of growth defects as body weight did not differ between genotypes (Figure 1—figure supplement 1D). To test if lethality occurs from gain-of-function TALK-1 L114P-mediated defect in neonatal islet function, we assessed glycemic control and insulin secretion on P4. Heterozygous and homozygous Kcnk16 L114P neonates showed severe hyperglycemia and a concurrent reduction in plasma insulin levels in a gene dosage-dependent manner compared to the control littermates (WT) on P4 (Figure 1C and D, and Figure 1—figure supplements 1E and 2A, B). Hyperglycemia in neonatal Kcnk16 L114P mice subsided by P10, when glycemia was equivalent to WT controls (Figure 1—figure supplement 2). Additionally, the Kcnk16 L114P mutation blunted glucose-stimulated insulin secretion in P4 islets from heterozygous neonates and decreased it further in P4 islets from homozygous neonates (Figure 1E and F). These changes in glucose-stimulated insulin secretion and glucose homeostasis are likely due to TALK-1 L114P channel mediated alteration in islet function and not islet mass as pancreas weight was unchanged (Figure 1G and Figure 1—figure supplement 1F). Furthermore, neonatal islet composition was unaltered except for a modest increase in the α-cell (glucagon+) area/islet in the Kcnk16 L114P (L/P) pancreas (Figure 1H–J). The loss of glucose-stimulated insulin secretion can be explained by a complete lack of glucose-stimulated Ca2+ entry in islets from Kcnk16 L114P (L/P and P/P) neonates (Figure 1K–N). However, these islets exhibited KCl-mediated β-cell Vm depolarization-induced Ca2+ entry; this indicates that TALK-1 L114P channels hyperpolarize β-cell Vm, thereby overriding the effect of KATP closure due to glucose metabolism and lead to insufficient glucose-stimulated insulin secretion and hyperglycemia. Importantly, insulin treatment (Glargine Lantus; 0.2 U/kg/day, once-daily subcutaneous injection) was able to extend the lifespan of Kcnk16 L114P (P/P) neonates, suggesting lethality results from hyperglycemia due to inadequate insulin secretion (Figure 1O).
Figure 1 with 2 supplements see all
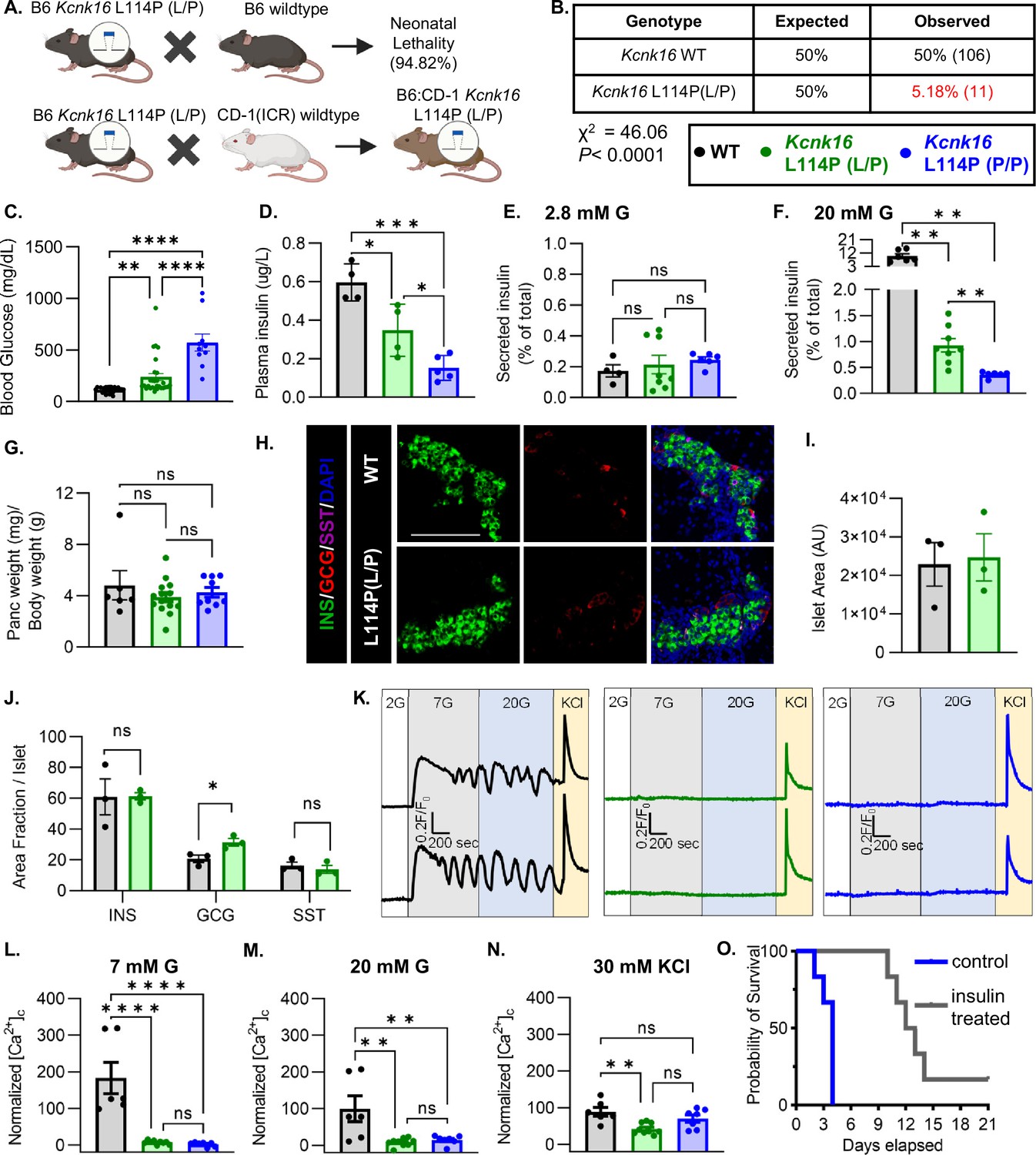
Kcnk16L114P neonates exhibit loss of glucose-stimulated Ca2+ entry and insulin secretion leading to transient neonatal hyperglycemia and death.
(A) Schematic of the Kcnk16 L114P mouse line generation in the C57BL/6J background and the C57BL/6J:CD-1 (ICR) mixed background. (B) χ2 analysis of the F1 progeny from C57BL/6J and heterozygous Kcnk16 L114P (L/P) crossings genotyped at weaning on postnatal day 21 (P21). (C) Blood glucose measurements of male wildtype (WT; black; N=8), heterozygous Kcnk16 L114P (L/P; green; N=16), and homozygous Kcnk16 L114P (P/P; blue; N=9) mice on P4. (D) Plasma insulin measurements performed on P4 in WT (N=4), Kcnk16 L114P (L/P; N=4), and Kcnk16 L114P (P/P; N=5) neonates. (E, F) In vitro glucose-stimulated insulin secretion from P4 mouse islets stimulated with 2.8 mM glucose (G) or 20 mM G, respectively (WT; N=4, Kcnk16 L114P (L/P); N=8, and Kcnk16 L114P (P/P); N=6). (G) Pancreas weight/ body weight measurements of P4 male mice (WT; N=6, Kcnk16 L114P (L/P); N=14, and Kcnk16 L114P (P/P); N=9). (H) Representative immunostaining images of pancreas sections from P0 WT and Kcnk16 L114P (L/P) mice (N=3 mice/genotype), stained against insulin (INS; green), glucagon (GCG; red), somatostatin (SST; magenta), and Hoechst (blue); scale bar = 100 μm. (I, J) Average islet area and area fraction of hormone staining per islet quantified using Fiji ImageJ software in P0 mouse pancreas sections. (K) Representative glucose-stimulated Ca2+ influx traces from P4 mouse islets sequentially stimulated with 2 mM glucose (G), 7 mM G, 20 mM G, and 20 mM G with 30 mM KCl (WT; N=6, Kcnk16 L114P (L/P); N=9, and Kcnk16 L114P (P/P); N=7). (L–N) Average area under the curve (AUC) analysis of normalized Ca2+ during 7 mM G, 20 mM G, and 20 mM G+30 mM KCl stimulations. (O) Survival curve for Kcnk16 L114P (P/P) mice treated with (N=6) or without (N=6) insulin (Lantus insulin glargine; 0.2 U/kg/day) starting at P0 up until death or weaning. Data are presented as mean ± SEM. Data were analyzed using Student’s t-test, one-way ANOVA, and two-way ANOVA as appropriate. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Adult Kcnk16 L114P mice exhibit fasting hyperglycemia and glucose intolerance
Because neonatal Kcnk16 L114P (L/P) mice showed transient hyperglycemia (Figure 1—figure supplement 2), we next utilized an intraperitoneal glucose tolerance test (GTT) to determine if Kcnk16 L114P impairs glucose homeostasis in young adulthood. Male Kcnk16 L114P (L/P) mice developed glucose intolerance as early as 11 weeks of age in the B6;CD-1 background and 8 weeks of age in the B6 background compared to their respective control littermates (data not shown). Similarly, male B6;CD-1 Kcnk16 L114P (L/P) mice developed impaired oral glucose tolerance by 7 weeks of age (Figure 2—figure supplement 1). This defect in glucose homeostasis is maintained with age in the male Kcnk16 L114P (L/P) mice (Figure 2A–C and Figure 2—figure supplements 1A–C and 2A–C). Furthermore, male Kcnk16 L114P (L/P) mice also exhibit fasting hyperglycemia indicating a likely non-β-cell-specific islet defect in this mouse model (Figure 2B). There were no body weight differences in males (Figure 2D). Female Kcnk16 L114P (L/P) mice only developed moderate glucose intolerance compared to the controls (WT) in both B6 and B6;CD-1 genetic backgrounds (Figure 2E–G; Figure 2—figure supplement 1D–F, 2E, F). Moreover, the Kcnk16 L114P mutation resulted in greater body weight gain in B6;CD-1 females compared to the WT controls (Figure 2H) due to increased lean mass, which might reduce glucose intolerance in Kcnk16 L114P (L/P) females compared to males (Figure 2—figure supplement 3A–F). The T2D-associated SNP in KCNK16 (rs1535500) which causes gain-of-function in TALK-1 activity has shown a significant association with increased total cholesterol in a Han Chinese population (Permana et al., 2019). However, an increase in TALK-1 activity due to the Kcnk16 L114P mutation did not affect plasma and liver triglyceride and cholesterol levels in mice (Figure 2—figure supplement 3G–J).
Figure 2 with 3 supplements see all
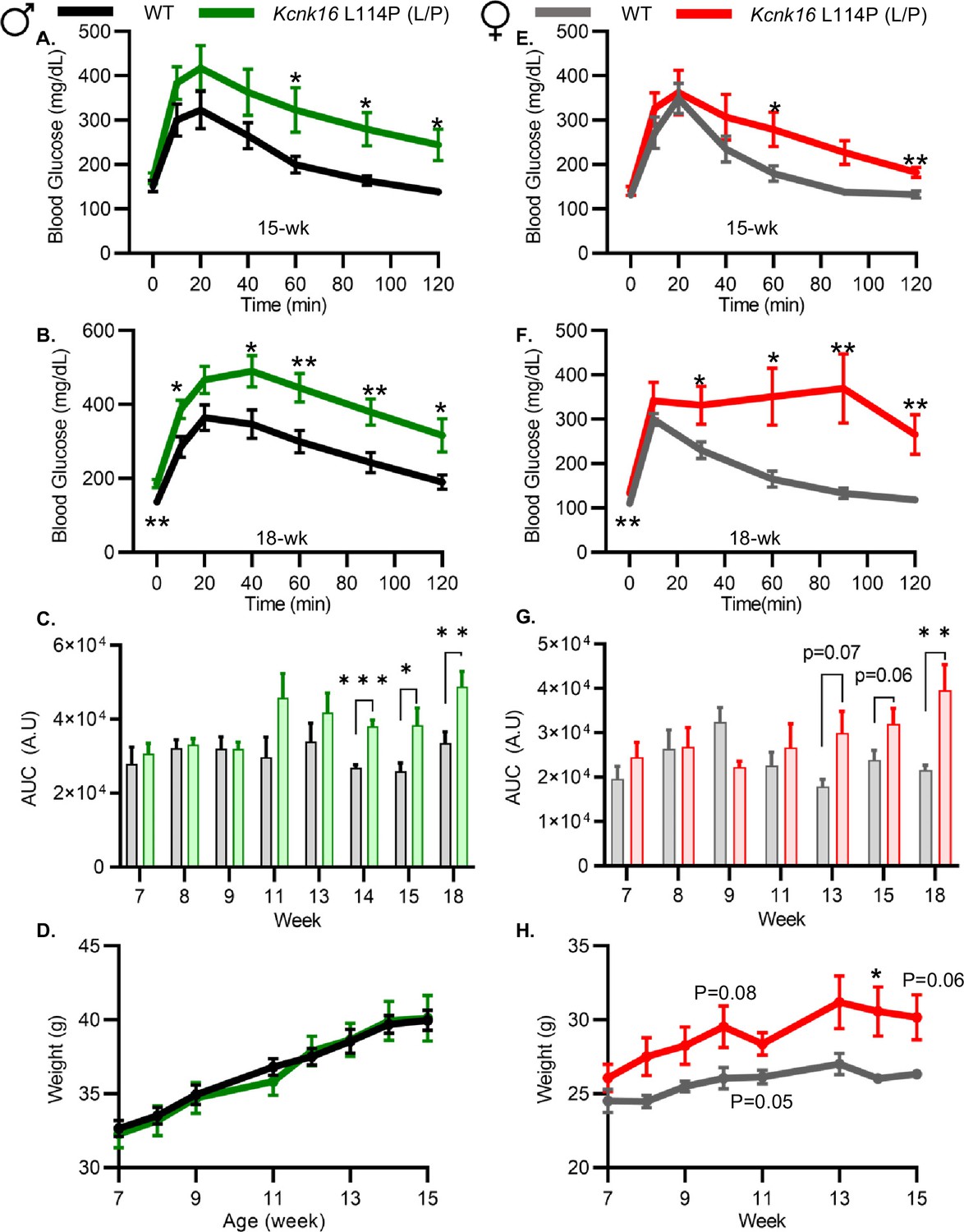
Adult Kcnk16 L114P mice exhibit fasting hyperglycemia and glucose intolerance.
(A, B) Intraperitoneal glucose tolerance test (i.p. GTT) performed in 15-week-old and 18-week-old male mice following a 4 hr fast in response to 2 mg/g glucose injection (WT; black; N=8–10 and Kcnk16 L114P (L/P); green; N=9–10). (C) Average area under the curve (AUC) of the 2 hr GTT excursion profiles from ages 7 weeks up to 18 weeks in male mice. (D) Weekly body weight measurements of male WT; N=5 mice and Kcnk16 L114P (L/P); N=5 mice. (E, F) I.p. GTT performed in 15-week-old and 18-week-old female mice following a 4 hr fast in response to 2 mg/g glucose injection (WT; N=9–11 and Kcnk16 L114P (L/P); N=10–11). (G) Average AUC of the 2 hr GTT excursion profiles from ages 7 weeks up to 18 weeks in female mice. (H) Body weight measurements of female WT (black; N=4) and Kcnk16 L114P (L/P; blue; N=5) mice. Data are presented as mean ± SEM. Data were analyzed using Student’s t-test. *p<0.05, **p<0.01, and ***p<0.001.
Adult Kcnk16 L114P mice show disrupted islet hormone secretion and islet composition
We then tested if the TALK-1-L114P-mediated impairment in glucose homeostasis arises from defective islet function. Both male and female Kcnk16 L114P (L/P) mice exhibited reduced plasma insulin levels compared to controls at 15 min and 30 min following an intraperitoneal glucose injection after a 4 hr fast (Figure 3A and B). Insulin sensitivity was not altered in the Kcnk16 L114P (L/P) mice indicating that the reduction in glucose tolerance is primarily due to an islet secretion defect (Figure 3C). This was confirmed by in vitro glucose-stimulated insulin secretion and glucose inhibition of glucagon secretion measurements (Figure 3D and E). Islets from Kcnk16 L114P (L/P) mice exhibited reduced ability to secrete insulin in response to glucose and showed elevated glucagon secretion under low glucose (2 mM) and euglycemic (7 mM) conditions (Figure 3D and E). This indicates that the fasting hyperglycemia observed in male Kcnk16 L114P (L/P) mice (Figure 2B) is likely a result of the observed increase in glucagon secretion under fasting conditions. We next investigated if impaired islet hormone secretion results from changes in islet composition. Immunostaining analyses revealed an increase in glucagon-positive area/islet and a concurrent modest reduction in insulin-positive area/islet in Kcnk16 L114P (L/P) pancreata (Figure 3F–J). Together, these results indicate that Kcnk16 L114P mutation leads to disruptions in both islet composition and secretion giving rise to a MODY-like phenotype in adult mice.
Figure 3
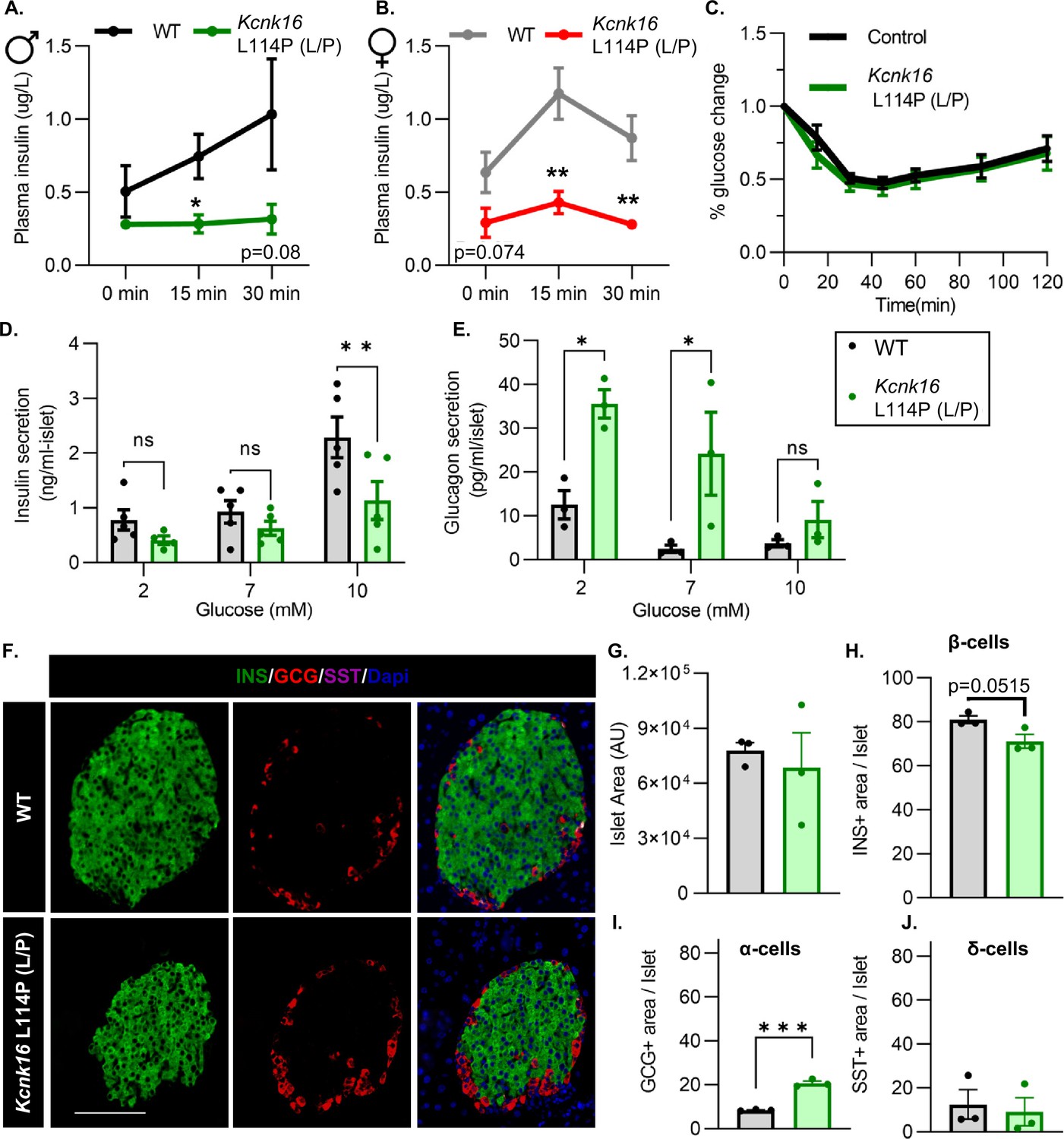
Adult Kcnk16 L114P mice show disrupted islet hormone secretion and islet composition.
(A, B) Plasma insulin levels in male (A) and female (B) WT and Kcnk16 L114P (L/P) mice following a 4 hr fast at the indicated time points before and after a 2 mg/g glucose injection. (C) Glucose (%) change in response to an intraperitoneal (i.p.) human insulin injection (0.75 UI/kg body weight) was measured as an indicator of insulin sensitivity in WT and Kcnk16 L114P male mice after a 4 hr fast. (D, E) In vitro insulin secretion (N=5 mice/genotype) and glucagon secretion (N=3 mice/genotype) from male mice at the specified glucose concentrations. (F) Representative immunostaining images of pancreas sections from WT and Kcnk16 L114P (L/P) male mice (N=3/genotype) stained against insulin (INS; green), glucagon (GCG; red), somatostatin (SST; magenta), and Hoechst (blue); scale bar = 100 μm. (G–J) Average islet area and area of hormone staining/islet for β-cells (INS), α-cells (GCG), and δ-cells (SST). Data are presented as mean ± SEM. Data were analyzed using Student’s t-test or two-way ANOVA as appropriate.*p<0.05, **p<0.01, and ***p<0.001.
Kcnk16 L114P blunts β-cell glucose-stimulated electrical excitability and increases whole-cell two-pore domain K+ channel currents
We used patch-clamp electrophysiology to test if the reduction in glucose-stimulated insulin secretion results from decreased β-cell electrical excitability due to a gain-of-function in TALK-1 activity in Kcnk16 L114P mouse islets. Perforated patch-clamp recordings revealed blunting of β-cell Vm depolarization and loss of action potential firing in islets from Kcnk16 L114P (L/P) mice (Figure 4A and B). Because the whole islet recordings assume β-cell electrical responses in response to glucose, perforated patch-clamp recordings were also run on islet cell clusters expressing a fluorescent reporter specifically in β-cells. Similar to the whole islet recordings, Kcnk16 L114P β-cells showed blunted glucose-stimulated Vm depolarization, which did not reach a depolarized threshold to fire action potentials in 67% of β-cells (Figure 4—figure supplement 1). However, as 33% of Kcnk16 L114P β-cells showed glucose-stimulated action potential firing (Figure 4—figure supplement 1), this indicates that these cells may also undergo Ca2+ entry and glucose-stimulated insulin secretion. Reduced glucose-stimulated islet Vm depolarization and action potential firing in Kcnk16 L114P β-cells was in part a result of increased whole-cell two-pore domain K+ channel currents at depolarized Vm and thus reducing K+ channel activity with high KCl depolarized these cells (Figure 4C and D; Figure 4—figure supplements 1 and 2). However, TALK-1 L114P (L/P and P/P) channels did not show a substantial increase at hyperpolarized β-cell membrane potentials under low and high glucose where these K+ currents are typically active (Figure 4—figure supplement 2). Notably, the heterologously expressed human TALK-1 L114P channels exhibited a drastic gain-of-function (Graff et al., 2021); this suggests the likelihood of unknown endogenous regulator(s) of TALK-1 in β-cells. It is also likely that recording conditions lead to poor isolation of the endogenous TALK-1 L114P currents. Thus, two-pore domain K+ channel currents were assessed by overexpressing mouse TALK-1 L114P channels in HEK293T cells under identical recording conditions as the human TALK-1 L114P study. Indeed, overexpression of the mouse mutant channel results in large K+ currents indicative of a similar gain-of-function in mouse TALK-1 current as the human TALK-1 current due to the L114P mutation (Figure 4E and F).
Figure 4 with 2 supplements see all
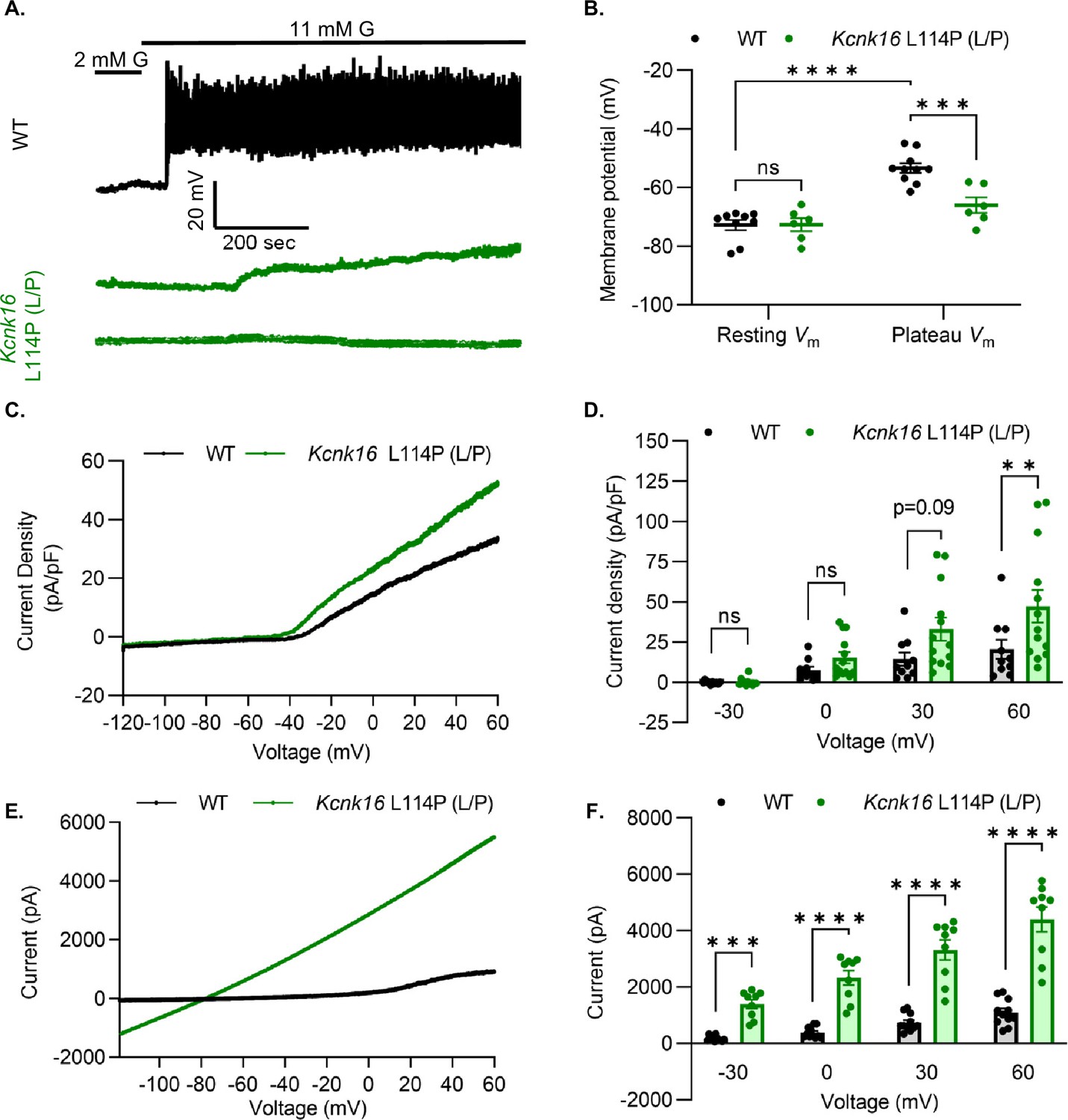
Kcnk16 L114P blunts β-cell glucose-stimulated electrical excitability and increases whole-cell two-pore domain K+ channel currents.
(A) Representative perforated patch-clamp Vm recordings in response to 2 mM G and 11 mM G in islets from WT and Kcnk16 L114P (L/P) mice (N=6–10 islets from 5 mice/genotype). (B) Average resting islet Vm under 2 mM G and plateau islet Vm at 11 mM G. (C) Representative whole-cell two-pore domain K+ channel current density (pA/pF) recorded using a voltage ramp (–120 mV to +60 mV) at 11 mM G in β-cells from WT and Kcnk16 L114P (L/P) mice. (D) Average current density (pA/pF) at the specified membrane voltages during the voltage ramp recordings shown in panel C (N=9–13 cells/genotype). (E) Representative whole-cell two-pore domain K+ channel current (pA) recorded using a voltage ramp (–120 mV to +60 mV) in 11 mM G in HEK293T cells expressing either Kcnk16 WT or Kcnk16 L114P (L/P). (F) Average current (pA) at the specified membrane voltages during the voltage ramp recordings shown in panel E (N=9–11 cells/ group). Data are presented as mean ± SEM. Data were analyzed using two-way ANOVA. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Kcnk16 L114P reduces glucose- and tolbutamide-stimulated Ca2+ entry and augments IP3-induced [Ca2+]ER release
β-Cell Ca2+ entry was monitored in response to increasing concentrations of glucose in islets from WT and Kcnk16 L114P (L/P) mice to assess if TALK-1 L114P-mediated Vm hyperpolarization reduces glucose-stimulated [Ca2+]c influx. TALK-1 L114P blunted islet glucose-stimulated [Ca2+]c influx in mice on both B6 and B6;CD-1 genetic backgrounds compared to WT controls (Figure 5A–F and Figure 5—figure supplement 1). However, islets from male Kcnk16 L114P (L/P) mice showed a larger reduction in normalized Ca2+ peak and normalized area under the curve responses compared to their controls than islets from female Kcnk16 L114P (L/P) mice compared to their controls (Figure 5A–F). TALK-1 L114P channels did not affect the basal Ca2+ levels at 2 mM G; however, the activity of these channels resulted in increased initial glucose-stimulated drop in [Ca2+]c (termed phase 0 response) compared to controls (Figure 5A; Figure 5—figure supplement 1). This suggests a reduction in endoplasmic reticulum (ER) Ca2+ ([Ca2+]ER) storage in Kcnk16 L114P islets. Furthermore, Kcnk16 L114P islets also showed a complete lack of glucose-stimulated [Ca2+]c oscillations monitored at 9 mM G (Figure 5—figure supplement 2C and D). We then assessed if the reduction in glucose-stimulated [Ca2+]c influx results from TALK-1 L114P overactivity on the β-cell plasma membrane by monitoring tolbutamide-stimulated Vm depolarization-induced [Ca2+]c entry. TALK-1 L114P-mediated β-cell Vm hyperpolarization overrides the Vm depolarization caused by KATP closure by tolbutamide. Thus, Kcnk16 L114P islets do not exhibit [Ca2+]c entry in response to tolbutamide. Interestingly, islets from Kcnk16 L114P mice showed equivalent KCl-stimulated [Ca2+]c entry compared to control islets; this suggests that the loss of glucose-stimulated [Ca2+]c influx in the Kcnk16 L114P islets occurs due to enhanced K+ ion flux through gain-of-function in TALK-1 activity (Figure 5G and H). Our previous studies showed that TALK-1 channels are functional on the ER membrane where they provide a K+ countercurrent for Ca2+ release from the ER lumen (Vierra et al., 2017). Thus, we tested if TALK-1 L114P regulates [Ca2+]ER homeostasis by stimulating Gq-signaling using acetylcholine, which results in IP3 generation and IP3-induced [Ca2+]ER release. Indeed, IP3-induced [Ca2+]ER release was enhanced in islets from Kcnk16 L114P mice compared to controls suggesting gain-of-function TALK-1 channels are localized to the ER membrane where they facilitate increased Ca2+ release from the ER lumen (Figure 5I and J).
Figure 5 with 2 supplements see all

Kcnk16 L114P reduces glucose- and tolbutamide-stimulated islet Ca2+ entry and augments IP3-induced islet [Ca2+]ER release.
(A, B) Representative [Ca2+]c traces in islets from male (A) and female (B) WT and Kcnk16 L114P (L/P) mice in response to 2 mM G, 10 mM G, and 20 mM G. (C–F) Average normalized Ca2+ peak (C and E) and total area under the curve (AUC) (D and F) in response to the indicated glucose concentrations in islets from male and female WT and Kcnk16 L114P (L/P) mice (N=3–4 mice/genotype). (G) Representative [Ca2+]c traces in islets from WT and Kcnk16 L114P (L/P) male mice in response to 100 μM tolbutamide followed by 30 mM KCl stimulation. (H) Average AUC of normalized [Ca2+]c during 100 μM tolbutamide or 100 μM tolbutamide with 30 mM KCl stimulation (N=3 mice/genotype). (I) Representative [Ca2+]c traces in response to 100 μM acetylcholine in the absence of extracellular Ca2+ in islets from male WT and Kcnk16 L114P (L/P) mice. (J) Average AUC of normalized [Ca2+]c following 100 μM acetylcholine-stimulated [Ca2+]ER release (N=876 cells; WT, N=513 cells; Kcnk16 L114P (L/P)). Data are presented as mean ± SEM. Data were analyzed using Student’s t-test. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Kcnk16 L114P islets exhibit altered expression of genes involved in β-cell identity, function, ion channel activity, inflammatory signaling, and extracellular matrix interaction pathways
Despite the loss of glucose-stimulated islet electrical activity and Ca2+ entry, Kcnk16 L114P mice do not exhibit a drastic reduction in glucose-stimulated insulin secretion and overt diabetes. Therefore, compensatory mechanisms such as the amplification pathway for insulin secretion might be altered in Kcnk16 L114P islets (Henquin, 2000). Moreover, prolonged hyperglycemia (although modest in this model) would also be predicted to result in long-term gene expression changes in Kcnk16 L114P islets that alter β-cell function. Thus, gene expression differences between WT and Kcnk16 L114P mouse islets were quantified with bulk RNA sequencing and validated with qRT-PCR (Figure 6A–D). We observed increased expression of many genes which regulate Ca2+-independent potentiation of insulin secretion such as Adcy5, Creb5, Adcyap1, and Adcyap1r1; genes that may promote Ca2+-independent secretion from Kcnk16 L114P islets (Hodson et al., 2014; Jamen et al., 2002). For example, Adcy5 has been shown to amplify glucose-stimulated cAMP production, which enhances glucose-stimulated insulin secretion. As cAMP is also a critical signal for β-cell Ca2+ entry (Capozzi et al., 2019; Zaborska et al., 2022), the impact of cAMP on Kcnk16 L114P islet Ca2+ handling was examined. Elevation of cAMP through activation of the GLP-1 receptor (with 200 nM liraglutide) only increased glucose-stimulated [Ca2+]c oscillation frequency in WT islets and did not impact the loss of glucose-stimulated [Ca2+]c entry in Kcnk16 L114P islet (Figure 6—figure supplement 1). Future studies are required to determine if and how gene expression changes promote Ca2+-independent glucose-stimulated insulin secretion in Kcnk16 L114P islets.
Figure 6 with 2 supplements see all
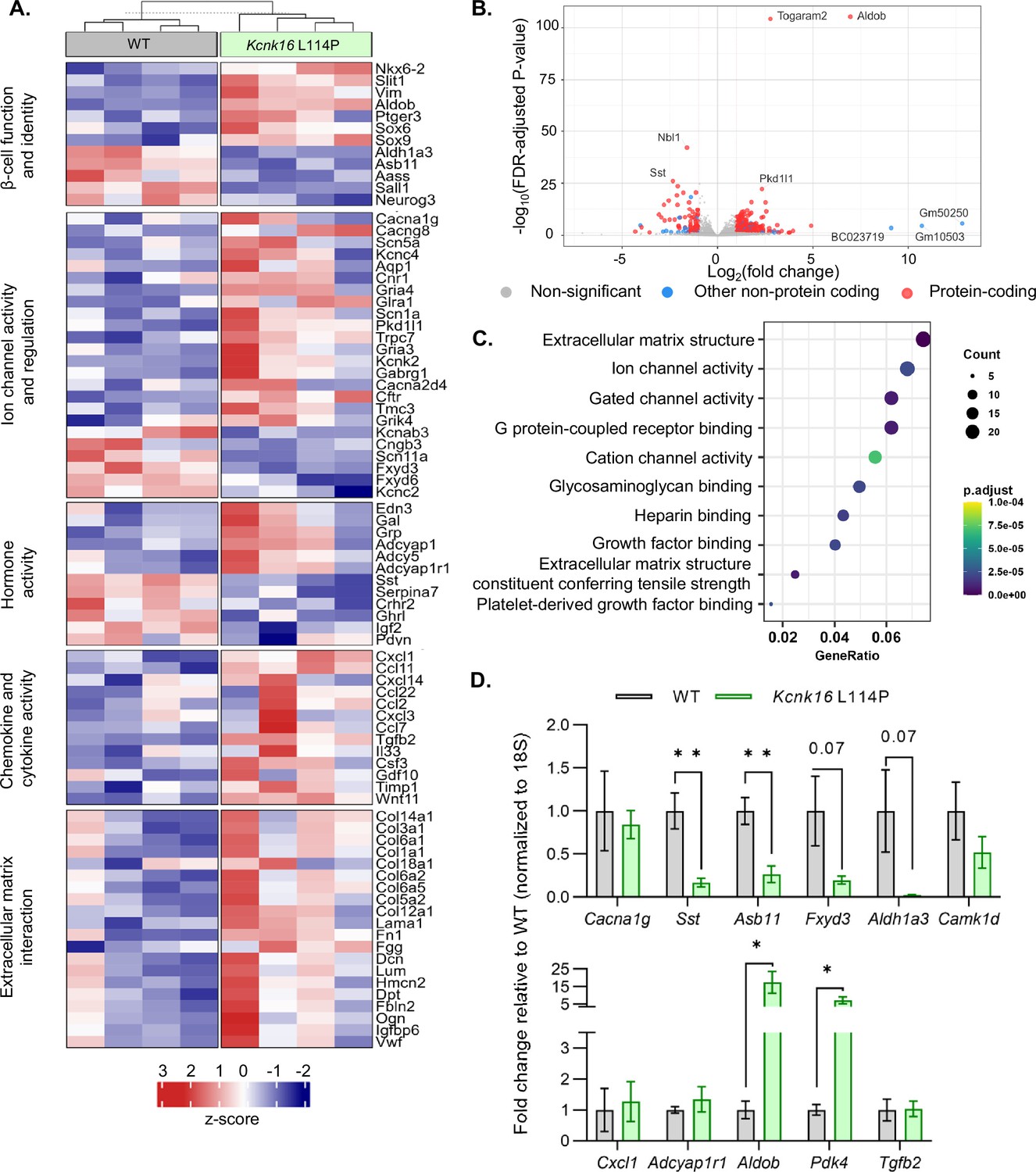
Kcnk16 L114P islets exhibit altered expression of genes involved in β-cell identity and function, ion channel activity, hormone activity, inflammatory signaling, and extracellular matrix interaction pathways.
(A) Heatmap of a selected gene subsets showing differential gene expression in WT and Kcnk16 L114P islets. Normalized expression levels were scaled and centered by rows. (B) Volcano plot displays genes differentially expressed between WT and Kcnk16 L114P samples. Differentially expressed genes are defined by FDR <0.05 and log2FC (≥1). (C) Dotplot represents the top 10 most significantly (FDR <0.05) altered Gene Ontology (Molecular Function). GeneRatio represent (count of enriched genes)/(count of genes in the GO term). The color represents FDR-adjusted p-values and the size of the dot represents the number of genes that are significant from the experimental dataset. (D) qRT-PCR validation of the gene expression differences in WT and Kcnk16 L114P samples for the selected genes observed through bulk RNA sequencing. *p<0.05, and **p<0.01.
Other pathways could also be impacted in Kcnk16 L114P islets that may modify Ca2+ handling or respond to the altered signaling such as in response to reduced glucose-stimulated [Ca2+]c influx, fasting hyperglycemia, and/or impaired glucose tolerance. For example, broad changes in ion channel activity genes (e.g. Cacna1g, Cacng8, Scn5a, Pkd1l1, Kcnk2, Gabrg1, and Fxyd3) may contribute to altered Ca2+ handling in Kcnk16 L114P islets. Reduced Ca2+ entry in Kcnk16 L114P islets results in decreased expression of genes previously shown to be elevated in chronically depolarized Abcc8−/− β-cells such as Serpina7, Asb11, Sall1, and Aldh1a3 (Figure 6A; Stancill et al., 2017). However, there were no expression differences in [Ca2+]ER handling genes such as Kcnk3, Itpr1, Itpr2, or Iptr3 (Figure 6—figure supplement 2). Furthermore, several stress-associated, fibrosis-related, and inflammatory signaling pathway genes are upregulated in Kcnk16 L114P islets likely due to prolonged hyperglycemic conditions. These include dedifferentiation markers Sox4, Sox6, Sox9, Hk2, Vim, and Cd36, extracellular matrix-interaction pathway genes Col1a1, Col1a2, Col3a1, Col14a1, Col6a1, and Dcn, and inflammatory signaling genes Cxcl1, Ccl2, Ccl11, Ccl22, Tgfb2, Il33, and Il6 (Roefs et al., 2017; Puri et al., 2013; Szabat et al., 2011; Eguchi and Nagai, 2017; Homo-Delarche et al., 2006; Hayden and Sowers, 2007). The data show that Kcnk16 L114P islets exhibit gene expression differences in many pathways. Taken together, this suggests that perturbed Kcnk16 L114P islet-intrinsic (e.g. Ca2+ handling) and -extrinsic (e.g. hyperglycemia) pathways result in direct as well as indirect disruption of islet function.
Discussion
Gain-of-function in TALK-1 activity is associated with diabetic phenotypes, suggesting a causal role for overactive TALK-1 in islet dysfunction and diabetes progression. Utilizing a gain-of-function model of TALK-1 (L114P), this study uncovered that disrupted β-cell and α-cell function resulted in glucose intolerance in adolescent mice confirming the association of the TALK-1 L114P mutation with MODY-like diabetes. Importantly, our data also revealed that Kcnk16 L114P mutation can cause transient neonatal diabetes. This finding suggests that transient neonatal diabetic patients with unknown genetic diagnosis should be screened for mutations in KCNK16. Moreover, our data provides further genetic evidence that TALK-1 is a potentially novel therapeutic target for diabetes treatment. Specifically, in mice heterozygous for Kcnk16 L114P mutation, we observe neonatal hyperglycemia due to blunted glucose-stimulated insulin secretion, which can additionally result in neonatal death in mice homozygous for the mutant (L114P) allele of Kcnk16. In young adulthood, Kcnk16 L114P causes glucose intolerance due to a reduction in glucose-stimulated insulin secretion mediated by enhanced β-cell Vm hyperpolarization and reduced glucose-stimulated Ca2+ entry. In addition to the β-cell-intrinsic defect, the Kcnk16 L114P mutation led to an increase in α-cell area fraction in islets and elevation of glucagon secretion under fasting conditions. Together these data highlight the crucial role of TALK-1 in β-cell function and glucose homeostasis and raises the possibility of TALK-1 inhibition as a druggable target for not only KCNK16-associated MODY but possibly for other forms of diabetes.
β-Cell maturation and glucose responsiveness of neonatal islets rapidly develops after birth and glucose-stimulated insulin secretion is required for efficient glucose uptake, which contributes to normal growth. Following birth, a shift to intermittent feeding and elevated plasma glucose requires an increase in β-cell insulin secretion for efficient nutrient absorption, as well as a suppression of insulin release during fasting to avoid hypoglycemia (Helman et al., 2020). Glucose sensitivity in mouse β-cell Ca2+ handling and insulin secretion develops over the first 4 postnatal days due to changes in β-cell metabolism, KATP surface localization, and Ca2+-dependent secretory machinery (West et al., 2021). Thus, mice expressing severe gain-of-function ATP-insensitive KATP channels die shortly after birth due to hypoinsulinemia, severe hyperglycemia, and ketoacidosis (Koster et al., 2000). Kcnk16 L114P mice show a similar phenotype, with neonatal islets showing a complete loss of glucose-stimulated [Ca2+]c influx, a drastic reduction in glucose-stimulated insulin secretion, and severe hyperglycemia by P4. This suggests a greater impact of Kcnk16 L114P in neonatal islets compared to adult islets. Future studies during β-cell maturation are required to determine if TALK-1 activity is greater on the plasma membrane and/or ER membrane in early neonatal compared with adult β-cells. Interestingly, Kcnk16 L114P-mediated susceptibility to neonatal lethality was dependent on genetic diversity in mouse strains. Heterozygous Kcnk16 L114P caused almost complete neonatal lethality in the B6 background but only hyperglycemia in the B6:CD-1 background, whereas homozygous Kcnk16 L114P led to neonatal lethality in the B6:CD-1 mice. The mechanism for the enhanced ability of heterozygous Kcnk16 L114P B6:CD-1 mice to survive remains to be determined. However, this has previously been reported for other knockout mouse models (e.g. Ankfy1 and Ywhaz), that are only viable as adults on a mixed genetic background (Weng et al., 2016; Yang et al., 2017). Although the neonatal glycemic data from the Kcnk16 L114P families does not exist, it may be that neonatal lethality is not observed in affected individuals. However, it is interesting to note that all currently identified individuals carrying the Kcnk16 L114P mutation are females (N=8) (Graff et al., 2021; Katsuyuki Matsui et al., 2023). The importance of hypoinsulinemia causing neonatal diabetes and lethality in TALK-1 L114P mice was confirmed by insulin treatment which extended their lifespan. This resembles other monogenic forms of neonatal diabetes that require exogenous insulin treatment for survival (Polak and Cavé, 2007). Indeed, it has been well established that insulin signaling is required for neonatal survival; for example, a similar neonatal lethality phenotype was observed in mice without insulin (Ins1/Ins2 double knockout) where death results within 2 days as well as in mice without insulin receptors (Insr-/-) where death results by P3 (Accili et al., 1996; Duvillié et al., 1997). Future studies are required to determine if TALK-1 gain-of-function mutations result in human transient neonatal diabetes followed by a MODY phenotype. Other monogenic diabetes mutations such as in genes encoding KATP channels result in transient neonatal diabetes with diabetes reemergence later in life (Flanagan et al., 2007).
In adolescence, the timeline of development of MODY-like diabetes in the Kcnk16 L114P (L/P) model is consistent with data from MODY patients. Similar to the timeline of disease progression in other MODY mouse models, Kcnk16 L114P mice developed glucose intolerance during adolescence (~8 weeks in the B6 strain). The onset and severity of glucose intolerance in Kcnk16 L114P mice also recapitulates the data from KCNK16 L114P MODY family probands, who were diagnosed at 11 and 15 years of age and displayed an abnormal oral GTT (blood glucose: 19 and 19.6 mM, 2 hr after 75 g oral glucose bolus, respectively). Disease severity is more prominent in the inbred B6 strain compared to the mixed B6:CD-1 strain likely owing to the outbred characteristics and genetic diversity of the CD-1 strain, which recapitulates the diverse nature of MODY manifestation observed in human patients. As the Japanese KCNK16 MODY family showed a greater insulin requirement compared to the patients from Australian KCNK16 MODY family, diversity in diabetes phenotypes likely occurs with KCNK16 mutations (Graff et al., 2021; Katsuyuki Matsui et al., 2023). Interestingly, male Kcnk16 L114P (L/P) mice exhibit more severe impairment in glucose homeostasis compared to female Kcnk16 L114P (L/P) mice. A variety of factors could explain the observed sex differences, including the female hormone 17β-estradiol (E2) which is critical for protection against glucolipotoxicity and oxidative stress (Gannon et al., 2018). Furthermore, older women have higher insulin levels in relation to relative insulin demand compared to men (Basu et al., 2017). Glucose-stimulated [Ca2+]c influx in islets from female Kcnk16 L114P mice is significantly greater than islets from male Kcnk16 L114P mice, which would be predicted to lead to greater insulin secretion and lesser impairment in glucose tolerance in female Kcnk16 L114P mice. Sexual dimorphism observed in glucose-stimulated [Ca2+]c influx could be mediated by differences in Ca2+ handling or differences in TALK-1 function. For example, under stress female human T2D islets maintain greater insulin secretion compared to males (Brownrigg et al., 2023). Although greater β-cell Kcnk16 expression could also be responsible for the more significant impairment in glucose intolerance observed in Kcnk16 L114P male mice, there is only a trend for greater Kcnk16 expression in sorted male β-cells (average RPKM 6296.25±953.84) compared to sorted female β-cells (5148.25±1013.22); this is similar in β-cells from high-fat diet (HFD)-treated mice (average RPKM 8020.75±1944.41 for males, and average RPKM 7551±2952.70 for females) (Brownrigg et al., 2023). Whether sex differences exist in humans with TALK-1 L114P mutation remains to be determined as all affected individuals in both KCNK16-MODY (p. TALK-1 L114P) families were females. Neonatal lethality was more penetrant in male than female Kcnk16 L114P (P/P) mice, thus, it will also be important to determine if KCNK16-MODY patients show neonatal phenotypes and display sexual dimorphism.
Similar to the proband from the human KCNK16 L114P MODY family who showed elevated fasting blood glucose (~7 mM), Kcnk16 L114P mice also exhibit fasting hyperglycemia. While this was predicted to be due to decreased insulin secretion, Kcnk16 L114P islets showed equivalent insulin secretion under euglycemic conditions. However, these islets exhibited a significant elevation in fasting glucagon secretion which likely contributes to the fasting hyperglycemia. If TALK-1 L114P channels were expressed in α-cells, it would result in inhibition of α-cell Ca2+ entry and glucagon secretion which supports previously described lack of TALK-1 protein expression in α-cells (Vierra et al., 2018). This suggests that hyperglucagonemia in TALK-1 L114P islets is likely due to loss of inhibitory paracrine signaling. As insulin secretion does not change under fasting and euglycemic conditions, hyperglucagonemia might primarily be mediated by reduced somatostatin secretion. This is supported by a significant decrease in Sst expression in Kcnk16 L114P islets (Figure 6), which is likely due to reduced δ-cell secretion. Also, our previous data in global TALK-1 KO mice show higher somatostatin secretion and lower glucagon secretion, thus δ-cell TALK-1 L114P would be predicted to limit Ca2+ influx and somatostatin secretion (Vierra et al., 2018). Intriguingly, Kcnk16 L114P islets additionally exhibit increased α-cell area fraction and a concurrent modest reduction in β-cell area fraction compared to control islets. These changes in islet composition is consistent with both T1D and T2D data showing increased α-cell:β-cell ratio (Li et al., 2000; Yoon et al., 2003), which show elevated islet glucagon secretion. α-Cell hyperplasia may result from increased activity/secretion, which is supported by other mediators of α-cell secretion (e.g. amino acids) that cause hyperplasia as well (Dean, 2020). However, loss of somatostatin also elevates α-cell secretion without altering α-cell mass (Hauge-Evans et al., 2009). Moreover, the increased somatostatin and insulin secretion only result in reduced α-cell secretion without altering α-cell number. Thus, the exact mechanism of how TALK-1 L114P mediates increased α-cell number remains to be determined. Taken together, the KcnK16 L114P mouse model shows disrupted glucagon and insulin secretion leading to fasting hyperglycemia and glucose intolerance, which provides confirmation that TALK-1 gain-of-function mutations likely cause MODY.
One obstacle in determining how MODY-associated mutations result in β-cell dysfunction is the limited availability of primary islet tissue from MODY families. Due to this, the initial assessment of MODY-associated human TALK-1 L114P was performed in overexpression systems, which resulted in a drastic gain-of-function. Similarly, overexpression of mouse Kcnk16 L114P leads to a substantial gain-of-function in TALK-1 activity (7.96-fold at –30 mV and 6.13-fold at 0 mV compared to TALK-1 WT). However, surprisingly β-cells from Kcnk16 L114P (L/P and P/P) mice showed only a modest gain-of-function in two-pore domain K+ channel currents. While the slight increase in β-cell K+ conductance from Kcnk16 L114P mice would be predicted to partially alter islet excitability, these islets exhibit a complete loss of glucose-stimulated Vm depolarization. This suggests that endogenous TALK-1 L114P polarizes plasma membrane potential, which is further supported by the robust KCl-induced Vm depolarization and Ca2+ entry observed in these islets. Because KCl shifts the reversal potential of K+ channels to a more depolarized Vm, the constant Vm hyperpolarization in TALK-1 L114P β-cells likely results from increased K+ conductance through these channels. Yet, K+ conductance in TALK-1 L114P β-cells was not significantly different at the membrane potentials of these cells under either low or high glucose conditions. The recording conditions may lead to poor isolation of the endogenous TALK-1 L114P currents; however, overexpression of this mutant channel results in large K+ currents under identical recording conditions. The K+ conductance differences between heterologously versus endogenously expressed TALK-1 L114P channels points toward unidentified regulators of β-cell TALK-1 activity which could include endogenous ligands, protein interactions, and cellular localization of the channel. This is presumably not due to changes in TALK-1 protein levels because Kcnk16 mRNA expression was not altered in control and Kcnk16 (c. 337T>C) islets. The subtle increase in β-cell K+ conductance correlates with a modest MODY-like phenotype and likely allows for incomplete suppression of β-cell function. This is also observed in KATP-MODY, where channel activity is only modestly increased (Yorifuji et al., 2005). Importantly, our study suggests that drastic TALK-1 gain-of-function mutations only lead to modest β-cell K+ conductance, which may explain why both families with TALK-1 MODY carry the same pore domain mutation (L114P) in TALK-1. Additionally, it is likely that other less-severe gain-of-function mutations in TALK-1 (e.g. A277E) result in a milder phenotype such as T2D. Another possibility is that because TALK-1 channels are functional not only on the plasma membrane but also the ER membrane (Vierra et al., 2017), some of the TALK-1 L114P-mediated reduction in glucose-stimulated [Ca2+]c influx could be due to altered [Ca2+]ER handling. Indeed, glucose-stimulated [Ca2+]c influx has previously been shown to be dependent in part on [Ca2+]ER release and TALK-1 L114P increases β-cell [Ca2+]ER release in response to muscarinic stimulation (Postic et al., 2023). Thus, future studies are required to determine the contributions of ER membrane localized versus plasma membrane localized TALK-1 L114P to altered glucose-stimulated [Ca2+]c influx. Moreover, it will be important to establish the contributions of endogenous modulators of TALK-1 channels (WT and L114P) and how they contribute to β-cell dysfunction.
Suppression of glucose-stimulated electrical activity and [Ca2+]c influx in Kcnk16 L114P islets would be predicted to cause a greater reduction of glucose-stimulated insulin secretion than that observed. However, a few β-cells from Kcnk16 L114P mice show modest Vm depolarization; this resulted in a slight increase in [Ca2+]c in response to glucose in a small subset of Kcnk16 L114P islets, which could result in some glucose-stimulated insulin secretion. Interestingly, Kcnk16 L114P islets also show reduced expression of Fxyd3, which encodes the auxiliary subunit of Na+/K+-ATPase and is a known negative regulator of glucose-stimulated insulin secretion in diabetic mice and humans (Vallois et al., 2014). Other interesting gene expression differences that may increase glucose-stimulated insulin secretion in Kcnk16 L114P islets include elevated expression (2.13-fold) of Adcy5, a Ca2+-independent amplification pathway gene (Hodson et al., 2014; Kalwat and Cobb, 2017). In humans, Adcy5 depletion impairs glucose-dependent elevation of cAMP and associated insulin secretion, thus elevation in Adcy5 expression would be predicted to increase glucose-stimulated insulin secretion via cAMP signaling (Hodson et al., 2014). Expression levels of other cAMP-dependent pathway genes were also elevated in the Kcnk16 L114P islets including Creb5, Adcyap1, and Adcyap1r1 (Jamen et al., 2002). Moreover, Slit1 and Srgap3, part of the SLIT-ROBO signaling which enhances glucose-stimulated insulin secretion, were upregulated (Yang et al., 2013). SLIT-ROBO signaling regulates not only Ca2+ handling but also actin remodeling and thus, Ca2+-independent signaling pathways. These gene expression changes suggest an increase in glucose-stimulated [Ca2+]c influx-independent mechanism(s) of insulin secretion may compensate for TALK-1 L114P-mediated loss of β-cell electrical activity and Ca2+ entry.
Chronic hyperglycemia in diabetic patients and rodents results in islet dysfunction and destruction, thus hyperglycemia observed in the Kcnk16 L114P mice could exacerbate β-cell failure. Glucotoxicity results in numerous islet transcriptome changes that in part contribute to dysfunction. Similar to islets from diabetic patients, Kcnk16 L114P islets exhibit an increase in Aldob (17.45-fold) and Nnat (1.95-fold) expression (Gerst et al., 2018; Millership et al., 2018). Additionally, Pdk4 expression, a marker for the shift from utilization of glucose to fatty acids as the primary fuel source, is higher in Kcnk16 L114P islets (2.78-fold). Elevated islet PDK4 expression is also observed in patients with T2D and in animals on an HFD (Eguchi and Nagai, 2017). Although hyperglycemia would be predicted to negatively impact Kcnk16 L114P islet function, no overt changes in β-cell mass were observed in these mice. This differs from mice with islets expressing KATP gain-of-function mutation, which show loss of β-cell mass (Shyr et al., 2019). Kcnk16 L114P may not cause a significant β-cell destruction because these mice show sufficient glucose-stimulated insulin secretion to prevent overt diabetes.
In summary, we showed that the MODY-associated TALK-1 L114P mutation elevates α-cell glucagon secretion under fasting and euglycemic conditions and blunts glucose-stimulated β-cell electrical activity and Ca2+ entry leading to reduced insulin secretion. Together, elevated glucagon impairs fasting glycemia and reduced glucose-stimulated insulin secretion increases post-prandial glucose levels in adults. This phenotype was more prominent in the male Kcnk16 L114P mice compared to the female Kcnk16 L114P mice raising the question whether sex differences translate in humans carrying this mutation. Surprisingly, the TALK-1 L114P mutation also resulted in severe transient neonatal diabetes which was lethal in the C57Bl/6J genetic background. Thus, these data hold potential clinical utility in that neonatal diabetes patients with unknown genetic linkage should be screened for mutations in KCNK16. Together, these data strengthen the rationale for designing TALK-1 inhibitors for use as a therapeutic modality to treat diabetes.
Limitations of the study: (1) This study does not establish how TALK-1 channels on the ER membrane influence glucose-stimulated [Ca2+]c influx. Future investigations are required to determine how Kcnk16 L114P modulation of β-cell [Ca2+]ER contributes to blunted glucose-stimulated [Ca2+]c influx and insulin secretion. (2) The Kcnk16 L114P mouse model created for this study replicates the MODY mutation in humans, however, due to TALK-1 expression in cells other than β-cells (e.g. δ-cells) it is difficult to determine the exact cellular contributions of Kcnk16 L114P to the MODY phenotype. (3) This study does not establish why TALK-1 L114P channel currents either from Kcnk16 L114P (L/P) or (P/P) primary β-cells show such low conductance compared to heterologously expressed TALK-1 L114P channels.
Methods
Chemicals and reagents
All research materials were purchased from Thermo Fisher (Waltham, MA, USA) or Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified.
Mouse model generation and ethical approval
Neonatal mice used for the studies were P0- to P5-old, and adult mice used for the studies were 6- to 26-week-old, age- and gender- matched, bred in-house on a C57BL/6J or mixed C57BL/6J:CD-1 (ICR) background. Animals were handled in compliance with guidelines approved by the Vanderbilt University Animal Care and Use Committee protocols (#M2200007-00). C57BL/6J.Kcnk16L114P mice (Kcnk16<em1Djaco>; MGI:7486559) were produced by the Vanderbilt Genome Editing Resource (Vanderbilt University, Nashville, TN, USA). Ribonucleoprotein complexes comprising chemically modified ctRNA (crRNA+tracrRNA) (50 ng/µl) and enhanced specificity SpCas9 protein (100 ng/µl), together with a 180-nucleotide single-stranded DNA (ssDNA) donor containing the Kcnk16 L114P mutation (50 ng/µl), were obtained from MilliporeSigma (Burlington, MA, USA). These components were diluted in 10 mM Tris, 0.1 mM EDTA, pH 7.6, sourced from Teknova (Half Moon Bay, CA, USA), and administered via pronuclear injection into C57BL/6J embryos acquired from mice from Jackson Labs (Bar Harbor, ME, USA). crRNA sequence: 5’CCCTGCAGGTTATGGAAACC. 180-Nucleotide ssDNA sequence: 5’ CTAGAGCTGGTGGTTGGGGGTGGGAGCCAGTTCTGGGCTCTCTTTTCCCCGCATCTGCACACTCCCTTGCCCTGCAGGTTATGGGAATCCAGCCCCCAGCACGGAGGCAGGGCAGGTCTTCTGTGTCTTCTATGCTCTGATGGGGATCCCACTCAATGTGGTCTTCCTCAACCATCTGGG. Mosaic F0 animals were screened for the L114P point mutation by standard PCR followed by a restriction fragment length polymorphism assay for a de novo HinfI site incorporated with silent mutations into the ssDNA. Animals carrying the desired mutation were confirmed by Sanger sequencing. Founder Kcnk16 L114P (L/P) mouse was backcrossed onto the C57Bl/6J (B6) strain for two generations to obtain mice used for all studies performed on the B6 background. Additionally, B6 Kcnk16 L114P (L/P) male mice were crossed with the CD-1 (ICR) strain to obtain F1 mice on a hybrid B6;CD-1 background to reduce the incidence of neonatal lethality. Crossings of heterozygous F1 B6;CD-1 Kcnk16 L114P (L/P) mice were used for generating homozygous B6;CD-1 Kcnk16 L114P (P/P) mice. For all the studies, littermates expressing the wildtype Kcnk16 allele were used as controls (WT).
Immunofluorescence
Mouse pancreata were fixed in 4% paraformaldehyde and embedded with paraffin. Rehydrated 5 μm sections were stained with primary antibodies against insulin (dilution 1:1000; Dako, Santa Clara, CA, USA), somatostatin (dilution 1:300, GeneTex., Irvine, CA, USA), and glucagon (dilution 1:100; Abcam, Cambridge, UK) followed by secondary antibodies (dilution 1:500; anti-guinea pig Alexa Fluor 488, dilution 1:500; anti-mouse, Alexa Fluor 647, and dilution 1:500; anti-rabbit Alexa Fluor 546) as previously described (Vierra et al., 2015). Sections were imaged either with a Nikon Eclipse TE2000-U microscope or fluorescent ScanScope (Aperio).
Islet isolation
Islets from neonatal mice were isolated on P4 using the protocol described by Huang and Gu, 2024. Briefly, pancreata were isolated and broken into ~2 mm pieces and digested in 200 µl collagenase P (Roche, Basel, Switzerland). For digestion, the tube was left in a 37°C incubator for up to 15 min and inverted two times every min. The lysate was spun at 500 × g for 10 s followed by three washes in RPMI, after which the islets were handpicked using a brightfield microscope.
Islets from adult mouse pancreata were isolated by collagenase P digestion and density gradient centrifugation as previously described (Vierra et al., 2015). Following isolation, islets were either dispersed into clusters of cells or single cells with trituration in 0.005% trypsin or maintained as whole islets. Cells were cultured in RPMI 1640 supplemented with 15% FBS, 100 IU/ml penicillin, 100 mg/ml streptomycin, and 5.5 mM glucose (RPMI) in a humidified incubator at 37°C with an atmosphere of 95% air and 5% CO2.
Whole-cell two-pore domain K+ channel currents
TALK-1 L114P currents were monitored using the whole-cell patch-clamp technique using an Axopatch 200B amplifier with pCLAMP10 software. Digidata 1440 was used to digitize currents that were low-pass-filtered at 1 kHz and sampled at 10 kHz. Cells were washed with the extracellular buffer (modified Krebs-Ringer-HEPES buffer [KRHB]) containing (mM) 119.0 NaCl, 2.0 CaCl2, 4.7 KCl, 25.0 HEPES, 1.2 MgSO4, 1.2 KH2PO4, and 11 mM glucose (pH 7.4 with NaOH). For isolation of two-pore domain K+ channel currents, KATP channels were blocked with 100 μM tolbutamide, voltage-gated K+ channels were blocked with 10 mM tetraethylammonium (Vierra et al., 2018; Vierra et al., 2017). Patch electrodes (3–5 MΩ) were backfilled with intracellular solution (IC) containing (mM) 140.0 KCl, 1.0 MgCl2, 10.0 EGTA, 10.0 HEPES, and 4.0 Mg-ATP (pH 7.25 with KOH). β-Cell Vm was ramped from –120 mV to +60 mV from a holding potential of –80 mV to generate two-pore domain K+ channel currents. Currents were measured in single β-cells from WT or Kcnk16 L114P (L/P) mice, or in HEK293FT cells (Catalog no. R70007, Invitrogen, Waltham, MA, USA) expressing either Kcnk16 WT or mouse Kcnk16 L114P channels. The whole-cell currents were analyzed using ClampFit (Molecular Devices) and Excel (Microsoft Corp., Redmond, WA, USA).
For two-pore domain K+ channel recordings in HEK293FT cells, the cells were grown to ~80% confluency in Dulbecco’s Modified Eagle Media (DMEM) GlutaMax-I (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Gibco), 100 IU/ml penicillin (Gibco), and 100 mg/ml streptomycin (Gibco) at 37°C, 5% CO2 in 100 mm tissue culture dishes. Cells were transfected with either pLV-CMV-mKcnk16:P2A:EGFP or pLV-CMV-mKcnk16 L114P:P2A:EGFP plasmids using Lipofectamine 3000 and P3000 (Thermo Fisher Scientific) in antibiotic-free Opti-MEM I Reduced Serum Medium as per the manufacturer’s protocol. Two-pore domain K+ channel currents were only recorded from EGFP-positive cells.
β-Cell Vm recordings
β-Cell Vm was recorded by the perforated patch-clamp technique using an Axopatch 200B amplifier with pCLAMP10 software on whole islets or islet clusters (containing 5–10 cells transduced with an adenoviral construct expressing GCaMP6s from a rat insulin promoter; Dickerson et al., 2022). Cells or islets were washed with KRHB with (mM) 119.0 NaCl, 2.0 CaCl2, 4.7 KCl, 25.0 HEPES, 1.2 MgSO4, 1.2 KH2PO4 (adjusted to pH 7.4 with NaOH) supplemented with 2 mM glucose and incubated in KRHB for 30 min at 37°C, 5% CO2. Patch electrodes (3–5 MΩ) were backfilled with IC containing (mM) 140.0 KCl, 1.0 MgCl2, and 5.0 HEPES (adjusted to pH 7.2 with KOH) supplemented with 20 μg/ml amphotericin B. For the islet clusters only β-cells expressing GCaMP6s were recorded. Islets and islet clusters were perifused with KRHB supplemented with 2 mM glucose followed by KRHB with 10 mM glucose for monitoring Vm changes. β-Cell Vm recordings were analyzed using ClampFit (Molecular Devices), Excel (Microsoft Corp., Redmond, WA, USA), and GraphPad Prism 8 (GraphPad Software Inc).
Intracellular Ca2+ imaging
On the day of experiment, islets were incubated for 30 min in RPMI supplemented with 2 μM Fura-2, AM (Molecular Probes) and 2 mM glucose. Fura-2, AM fluorescence (Ratio 340Ex/380Ex-535Em; F340/F380) was measured every 5 s as an indicator of intracellular Ca2+ using a Nikon Eclipse Ti2 microscope equipped with a Photometrics Prime 95B 25 mm sCMOS Camera (Zaborska et al., 2020). For [Ca2+]c measurements, β-cell glucose-stimulated Ca2+ influx was monitored in KRHB supplemented with the glucose concentrations specified in the figures. For IP3-induced [Ca2+]ER release measurements, islets were perifused in KRHB buffer containing 11 mM glucose, 100 μM diazoxide, without extracellular Ca2+. Fura-2, AM fluorescence was monitored as an indicator of IP3-mediated [Ca2+]ER release upon stimulation of muscarinic receptor signaling by 100 μM acetylcholine. For all measurements, the cells were perifused at a flow rate of 2 ml/min. Ex; excitation wavelength (nm), Em; emission wavelength (nm).
Glucose homeostasis
Chow-diet fed male and female mice underwent GTT and insulin tolerance test (ITT) as previously described (Vierra et al., 2015). Briefly, mice were fasted for 4 hr and then 2 mg dextrose/g body weight was administered with either intraperitoneal injection or oral gavage for GTTs, or 0.75 UI human recombinant insulin/kg body weight for ITTs (Catalog no. 12585014, Gibco). Tail glucose measurements were then taken at the indicated time points in the figures to measure glucose clearance.
Body composition, tissue and plasma triglyceride, and cholesterol measurements
Measurement of lean tissue, fat, and fluid in living mice was performed using Bruker’s minispec Body Composition Analyzer. Plasma samples and livers were collected from ad lib fed mice for triglyceride and cholesterol measurements. Total cholesterol and triglycerides were measured using standard enzymatic assays by the Vanderbilt University Medical Center Lipid Core.
Plasma insulin, in vitro insulin, and glucagon secretion assays
Plasma insulin from the neonates was measured on P4 using Ultrasensitive Mouse Insulin ELISA kits (Catalog no. 10-1249-01). In adult mice, plasma insulin measurements were conducted in mice fasted for ~4 hr followed by intraperitoneal injection of 2 mg dextrose/g body weight at 0, 15, and 30 min post injection. Tail blood samples were collected in Microvette CB 300 K2 EDTA tubes (Catalog no. 16.444.100, Sarstedt) at the indicated time points and plasma insulin were measured using mouse ultrasensitive insulin ELISA kits (Mercodia Inc, Sweden). For in vitro insulin and glucagon secretion assays, islets were isolated from mice fed a standard chow diet and were incubated overnight in RPMI supplemented with 0.5 mg/ml BSA. On the following day, islets were equilibrated in DMEM containing 0.5 mg/ml BSA, 0.5 mM CaCl2 and 10.0 mM HEPES (DMEM*) supplemented with 10% FBS and 5.5 mM glucose for 1 hr at 37°C, 5% CO2. 20 islets/well were picked into 400 µl DMEM* without FBS at glucose concentrations specified in the figures in 24-well plate(s) and insulin or glucagon secretion was measured over 1 hr at 37°C and stored at –20°C until analysis. Insulin secretion was measured using mouse insulin ELISA kits (Catalog no. 10-1247-01, Mercodia Inc, Sweden) and glucagon secretion was measured using mouse glucagon ELISA kits (Catalog no. 10-1281-01, Mercodia Inc, Sweden).
Bulk RNA sequencing
RNA was isolated from islets from ~15-week-old male mice (WT and Kcnk16 L114P (L/P)) using Maxwell 16 LEV simplyRNA Purification Kits (Catalog no. AS1280, Promega, USA). RNA integrity was analyzed using an Agilent 2100 Bioanalyzer, and only those samples with an RNA integrity number of seven or above were used. An Illumina NovaSeq 6000 instrument was used to produce paired-end, 150-nucleotide reads for each RNA sample. Paired-end RNA sequencing reads (150 bp long) were trimmed and filtered for quality using Trimgalore v0.6.7 (Felix Krueger et al., 2023). Trimmed reads were aligned and counted using Spliced Transcripts Alignment to a Reference (STAR) (Dobin et al., 2013) v2.7.9a with the –quantMode GeneCounts parameter against the mm10 mouse genome and GENCODE comprehensive gene annotations (Release M23). Per sample, the number of mapped reads ranged from 52 to 462 million. DESeq2 package v1.34.0 (Love et al., 2014) was used to perform normalization and downstream differential expression. Features counted fewer than five times across at least three samples were removed. Freezing condition of the samples and sequencing batch was included as batch factor in DESeq2 design to increase the sensitivity for finding differences between Kcnk16 L114P v/s WT samples and confirmed with principle component analysis (Figure 6—figure supplement 2B). Gene enrichment analysis implemented from Gene Ontology (GO) was applied using the clusterProfiler v4.2.2 package in R. Annotated gene sets GO was sourced from Genome wide annotation for Mouse (Carlson, 2019). For GO, genes significantly up- or downregulated in different conditions were used as input. False discovery rate-adjusted p-value <0.05 and log2 fold change >1 was used to define differentially expressed genes.
Quantitative PCR
RNA was isolated from islets from 15-week-old, chow diet fed mice using Maxwell 16 LEV simplyRNA Purification Kits (Catalog no. AS1280, Promega, USA). Reverse transcription was performed using a SuperScript IV First-Strand Synthesis System (Catalog no. 18091050, Invitrogen, Waltham, MA, USA). 20 ng cDNA was used for real-time qPCRs with KAPA SYBR FAST qPCR Kit (Catalog no. KK4618, Roche, Basel, Switzerland) using a CFX Opus Real-Time PCR System (Bio-Rad Laboratories). Primers used for qRT-PCR are listed in Supplementary file 1.
Statistical analysis
Functional data were analyzed using Axon Clampfit (Molecular Devices), GraphPad Prism 8 (GraphPad Software Inc), or Excel (Microsoft Corp., Redmond, WA, USA) and presented as mean ± standard error (SE) for the specified number of samples (N). Statistical significance was determined using two-tailed t-tests, one-way ANOVA, or two-way ANOVA as appropriate. p-Value ≤0.05 was considered statistically significant.
Data availability
RNA-sequencing data has been deposited and is available in NCBI GEO with accession ID GSE239566.
-
NCBI Gene Expression OmnibusID GSE239566. The MODY-associated TALK-1 L114P mutation causes islet α-cell overactivity and β-cell inactivity resulting in transient neonatal diabetes and glucose dyshomeostasis in adults.
References
-
Men are from mars, women are from venus: sex differences in insulin action and secretionAdvances in Experimental Medicine and Biology 1043:53–64.https://doi.org/10.1007/978-3-319-70178-3_4
-
Insight on diagnosis and treatment from over a decade of research through the university of chicago monogenic diabetes registryFrontiers in Clinical Diabetes and Healthcare 2:735548.https://doi.org/10.3389/fcdhc.2021.735548
-
Epigenomic plasticity enables human pancreatic α to β cell reprogrammingThe Journal of Clinical Investigation 123:1275–1284.https://doi.org/10.1172/JCI66514
-
Approach to the patient with MODY-Monogenic DiabetesThe Journal of Clinical Endocrinology & Metabolism 106:237–250.https://doi.org/10.1210/clinem/dgaa710
-
Sex differences in islet stress responses support female β cell resilienceMolecular Metabolism 69:101678.https://doi.org/10.1016/j.molmet.2023.101678
-
Non classic presentations of a genetic mutation typically associated with transient neonatal diabetesEndocrinology, Diabetes & Metabolism Case Reports 2020:19-0125.https://doi.org/10.1530/EDM-19-0125
-
STAR: ultrafast universal RNA-seq alignerBioinformatics 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Islet inflammation in type 2 diabetes and physiologyThe Journal of Clinical Investigation 127:14–23.https://doi.org/10.1172/JCI88877
-
Sex differences underlying pancreatic islet biology and its dysfunctionMolecular Metabolism 15:82–91.https://doi.org/10.1016/j.molmet.2018.05.017
-
The Expression of Aldolase B in islets is negatively associated with insulin secretion in humansThe Journal of Clinical Endocrinology and Metabolism 103:4373–4383.https://doi.org/10.1210/jc.2018-00791
-
Genomic and functional characteristics of novel human pancreatic 2P domain K(+) channelsBiochemical and Biophysical Research Communications 282:249–256.https://doi.org/10.1006/bbrc.2001.4562
-
Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetesThe New England Journal of Medicine 350:1838–1849.https://doi.org/10.1056/NEJMoa032922
-
Effective isolation of functional islets from neonatal mouse pancreasJournal of Visualized Experiments 2017:119.https://doi.org/10.3791/55160-v
-
Mechanisms of the amplifying pathway of insulin secretion in the β cellPharmacology & Therapeutics 179:17–30.https://doi.org/10.1016/j.pharmthera.2017.05.003
-
Single-channel properties and pH sensitivity of two-pore domain K+ channels of the TALK familyBiochemical and Biophysical Research Communications 315:836–844.https://doi.org/10.1016/j.bbrc.2004.01.137
-
ConferenceClinical presentation of Japanese patients with KCNK16-MODYInternational Meeting in Pediatric Endocrinology (IMPE.
-
Maturity onset diabetes of the young: clinical characteristics, diagnosis and managementPediatric Endocrinology Reviews 10:234–242.
-
Neuronatin regulates pancreatic β cell insulin content and secretionThe Journal of Clinical Investigation 128:3369–3381.https://doi.org/10.1172/JCI120115
-
Neonatal diabetes mellitus: a disease linked to multiple mechanismsOrphanet Journal of Rare Diseases 2:12.https://doi.org/10.1186/1750-1172-2-12
-
High-resolution analysis of the cytosolic Ca(2+) events in beta cell collectives in situAmerican Journal of Physiology. Endocrinology and Metabolism 324:E42–E55.https://doi.org/10.1152/ajpendo.00165.2022
-
Increased vimentin in human α- and β-cells in type 2 diabetesThe Journal of Endocrinology 233:217–227.https://doi.org/10.1530/JOE-16-0588
-
Pancreatic β-cell electrical activity and insulin secretion: of mice and menPhysiological Reviews 98:117–214.https://doi.org/10.1152/physrev.00008.2017
-
Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.KPediatric Diabetes Population With Monogenic Diabetes. Diabetes Care 39:1879–1888.https://doi.org/10.2337/dc16-0645
-
Ankfy1 is dispensable for neural stem/precursor cell developmentNeural Regeneration Research 11:1804–1809.https://doi.org/10.4103/1673-5374.194750
-
Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in KoreaThe Journal of Clinical Endocrinology and Metabolism 88:2300–2308.https://doi.org/10.1210/jc.2002-020735
-
The C42R mutation in the Kir6.2 (KCNJ11) gene as a cause of transient neonatal diabetes, childhood diabetes, or later-onset, apparently type 2 diabetes mellitusThe Journal of Clinical Endocrinology and Metabolism 90:3174–3178.https://doi.org/10.1210/jc.2005-0096
-
Monogenic diabetes: a gateway to precision medicine in diabetesThe Journal of Clinical Investigation 131:e142244.https://doi.org/10.1172/JCI142244
Article and author information
Author details
Funding
National Institutes of Health (R01DK097392)
- Prasanna K Dadi
- Matthew T Dickerson
- Soma Behera
- David A Jacobson
National Institutes of Health (R01DK129340)
- Arya Y Nakhe
- Prasanna K Dadi
- Matthew T Dickerson
- David A Jacobson
National Institutes of Health (R01DK115620)
- Arya Y Nakhe
- Prasanna K Dadi
- Matthew T Dickerson
- Jordyn R Dobson
- David A Jacobson
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The Kcnk16 L114P mouse model was developed by the Vanderbilt Genome Editing Resource Core (RRID:SCR018826) which is supported by the Diabetes Research and Training Center Grant (DK020593), and the Cancer Center Support Grant (CA68485), and the Vanderbilt Center for Stem Cell Biology. Paraffin-embedded pancreata were prepared and processed by The Vanderbilt Translational Pathology Shared Resource, which was supported by the Cancer Center Support Grant CA68485. Immunostaining slide scanning was performed using the Islet and Pancreas Analysis (IPA) Core supported by the Vanderbilt Diabetes Research Center (DRTC; NIH grant DK20593). Analysis of the bulk RNA sequencing data was performed by Creative Data Solutions, part of the Vanderbilt Center for Stem Cell Biology. Body composition measurements were performed at the Vanderbilt Mouse Metabolic Phenotyping Center which is supported by the NIH grant 5U2CDK059637. Triglyceride and cholesterol measurements were performed by the Vanderbilt University Medical Center Lipid Core supported by the NIH DRTC grant DK020593. Research in the laboratory of DAJ was supported by NIH grants R01DK097392, R01DK129340, and R01DK115620.
Ethics
Animals were handled in compliance with guidelines approved by the Vanderbilt University Animal Care and Use Committee protocols (#M2200007-00).
Version history
- Sent for peer review:
- Preprint posted:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.89967. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2023, Nakhe et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,422
- views
-
- 85
- downloads
-
- 6
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 4
- citations for umbrella DOI https://doi.org/10.7554/eLife.89967
-
- 1
- citation for Reviewed Preprint v1 https://doi.org/10.7554/eLife.89967.1
-
- 1
- citation for Version of Record https://doi.org/10.7554/eLife.89967.3
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The MODY-associated KCNK16 L114P mutation increases islet glucagon secretion and limits insulin secretion resulting in transient neonatal diabetes and glucose dyshomeostasis in adults
eLife 12:RP89967.
https://doi.org/10.7554/eLife.89967.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}